Python基因组处理,Biopython入门指南
Python处理基因组数据,Biopython是你的得力助手!本教程带你快速入门Biopython,掌握处理基因组数据的核心技能。首先,理解Seq和SeqRecord的区别至关重要,Seq代表序列本身,而SeqRecord则封装了序列及其元数据,如ID、名称和描述。其次,利用Bio.SeqIO模块,轻松读写FASTA和GenBank等常见基因组文件格式,parse()函数助你高效处理大型文件。最后,Biopython提供丰富的序列操作功能,包括转录、反向互补、翻译以及GC含量计算等,助你高效完成生物信息学分析任务。掌握Biopython,让基因组数据处理变得更简单、更高效!
Biopython的核心数据结构是Seq和SeqRecord。Seq表示DNA、RNA或蛋白质序列本身,包含碱基或氨基酸字符串及可选的字母表;SeqRecord则封装Seq对象,并附加id、name、description、features和annotations等元数据,代表一条完整的生物学记录。理解这两者的区别与联系,是掌握Biopython的关键。此外,Biopython通过Bio.SeqIO模块支持多种基因组文件格式的读写操作,如FASTA和GenBank,使用parse()逐条读取大文件以节省内存,用write()将序列记录导出为指定格式。在序列分析方面,Biopython提供丰富的内置功能,包括转录、反向互补、翻译(支持不同遗传密码子表)、GC含量计算、子序列查找及切片操作等,极大提升了生物信息学工作的效率与准确性。

Python,特别是Biopython库,是处理基因组数据的强大工具,它简化了序列操作、文件解析、比对等复杂任务,让生物信息学分析变得更加高效和直观。

我记得刚开始接触生物信息学时,面对那些庞大的基因组文件,感觉就像掉进了数据汪洋,不知从何下手。那时候,手动处理简直是噩梦。直到我遇到了Python和Biopython,它就像是给我递来了一艘小船,让我能在这片汪洋中找到方向,甚至开始享受探索的乐趣。

Biopython的核心在于它提供了一套直观的接口来表示生物学概念,比如序列、序列记录和比对。它不是一个包罗万象的分析套件,更像是一套积木,让你能用代码搭建自己的生物信息学工作流程。从最基础的读取FASTA文件,到执行复杂的序列比对,甚至是与NCBI等在线数据库交互,Biopython都能提供底层支持。
它让数据处理变得可编程、可重复,这对于任何科研工作来说都至关重要。你不再需要每次都点击图形界面,而是可以通过编写脚本,自动化那些重复性高、容易出错的任务。这不仅节省了大量时间,也大大提高了分析的准确性和一致性。

Biopython的核心数据结构是什么?如何理解序列和记录?
初学者常常会混淆Biopython中的Seq和SeqRecord,我当初也是。简单来说,Seq就是DNA、RNA或蛋白质链本身,它只包含了碱基或氨基酸的序列信息,以及一个可选的字母表(alphabet)来指明它是DNA、RNA还是蛋白质。比如,Seq("ATGC")就是一个DNA序列。这个设计很精妙,因为它提醒我们,序列本身可能没有太多元数据,它就是一串字符。
而SeqRecord则像是给这条链贴上了标签和说明书。它封装了一个Seq对象,并额外提供了id(唯一标识符)、name(名称)、description(描述)、features(特征,如基因、CDS区域等)和annotations(其他注释信息,如物种、来源等)。可以把它想象成数据库里的一条记录,包含了序列以及所有相关的元数据。在实际工作中,我们更多地是操作SeqRecord对象,因为它承载了分析所需的所有上下文信息。
例如,从FASTA文件读取时,每一条记录通常都会被解析成一个SeqRecord对象,其id可能就是FASTA头部的标识符。理解这两者的区别和联系,是掌握Biopython的第一步。
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.Alphabet import IUPAC
# 创建一个Seq对象
my_dna = Seq("ATGCGTACGTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGC", IUPAC.unambiguous_dna)
print(f"Seq对象: {my_dna}")
print(f"序列长度: {len(my_dna)}")
print(f"序列类型: {my_dna.alphabet}")
# 创建一个SeqRecord对象
record = SeqRecord(
my_dna,
id="NC_000913.1",
name="E.coli_K12",
description="Escherichia coli str. K-12 substr. MG1655, complete genome"
)
print(f"\nSeqRecord ID: {record.id}")
print(f"SeqRecord Name: {record.name}")
print(f"SeqRecord Description: {record.description}")
print(f"SeqRecord 包含的序列: {record.seq}")如何使用Biopython读取和写入常见的基因组文件格式?
处理基因组数据,第一步往往就是文件读写。我个人觉得Bio.SeqIO简直是神器,它抽象了各种文件格式的差异,让你可以用几乎一套代码搞定FASTA、GenBank这些常见的格式。无论是需要读取一个巨大的FASTA文件,还是将处理后的序列保存为新的GenBank格式,Bio.SeqIO都能轻松应对。
读取文件通常使用Bio.SeqIO.parse()函数,它会返回一个迭代器,这对于处理非常大的文件尤其有用,因为它不会一次性将所有数据加载到内存中。你可以逐条处理序列记录,避免内存溢出的问题。而写入文件则使用Bio.SeqIO.write()函数,它接受一个序列记录列表或迭代器,以及输出文件句柄和格式。
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.Alphabet import IUPAC
# 示例:创建一个简单的FASTA文件用于读取
with open("example.fasta", "w") as f:
f.write(">gene1 description_of_gene1\n")
f.write("ATGCGTACGTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGC\n")
f.write(">gene2 description_of_gene2\n")
f.write("GTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGC\n")
# 读取FASTA文件
print("--- 读取FASTA文件 ---")
for record in SeqIO.parse("example.fasta", "fasta"):
print(f"ID: {record.id}")
print(f"Description: {record.description}")
print(f"Sequence (first 20bp): {record.seq[:20]}...")
print("-" * 20)
# 示例:创建一些SeqRecord对象用于写入
records_to_write = [
SeqRecord(Seq("ATGCATGC", IUPAC.unambiguous_dna), id="seq_a", description="First sequence"),
SeqRecord(Seq("GCTAGCTA", IUPAC.unambiguous_dna), id="seq_b", description="Second sequence")
]
# 写入为新的FASTA文件
print("\n--- 写入新的FASTA文件 ---")
output_fasta_file = "output.fasta"
with open(output_fasta_file, "w") as output_handle:
SeqIO.write(records_to_write, output_handle, "fasta")
print(f"序列已写入到 {output_fasta_file}")
# 也可以写入GenBank格式(如果SeqRecord有足够信息)
# 注意:写入GenBank通常需要更完整的SeqRecord信息,如特征(features)
# output_genbank_file = "output.gb"
# with open(output_genbank_file, "w") as output_handle:
# SeqIO.write(records_to_write, output_handle, "genbank")
# print(f"序列已写入到 {output_genbank_file}")Biopython在序列操作和基本分析中有哪些实用功能?
拿到序列后,我们自然会想做些操作,比如看看它的互补链,或者把它翻译成蛋白质。Biopython在这方面提供了很多开箱即用的功能,省去了我们自己写那些繁琐的字符串处理代码。它内置了转录、反向互补、翻译等常用功能,并且考虑到了不同遗传密码子表的情况。
我记得有次调试一个转录翻译的脚本,结果发现输出总是错的,后来才发现是自己没注意Seq对象的alphabet属性,Biopython的严谨性有时候也会给我带来一些小“惊喜”,但更多的是保障了数据的准确性。此外,计算GC含量、查找特定模式等基础分析功能也一应俱全。
from Bio.Seq import Seq
from Bio.Alphabet import IUPAC
dna_seq = Seq("ATGCGTACGTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGC", IUPAC.unambiguous_dna)
# 1. 转录 (DNA -> RNA)
rna_seq = dna_seq.transcribe()
print(f"原始DNA: {dna_seq}")
print(f"转录RNA: {rna_seq}")
# 2. 反向互补
rev_comp_dna = dna_seq.reverse_complement()
print(f"反向互补DNA: {rev_comp_dna}")
# 3. 翻译 (RNA -> 蛋白质)
# 注意:直接对DNA序列进行翻译,Biopython会先进行转录
protein_seq = dna_seq.translate()
print(f"翻译蛋白质: {protein_seq}")
# 翻译时指定不同的遗传密码子表(例如:线粒体密码子)
# protein_seq_mito = dna_seq.translate(table="Vertebrate Mitochondrial")
# print(f"线粒体蛋白质: {protein_seq_mito}")
# 4. 计算GC含量
gc_content = (dna_seq.count("G") + dna_seq.count("C")) / len(dna_seq) * 100
print(f"GC含量: {gc_content:.2f}%")
# 5. 查找子序列(Python字符串方法)
# Biopython的Seq对象继承了Python字符串的大部分方法
if "ATGC" in dna_seq:
print("序列中包含 'ATGC'")
print(f"首次出现'TAGC'的位置: {dna_seq.find('TAGC')}") # 如果找不到返回-1
# 6. 切片操作
sliced_seq = dna_seq[10:20]
print(f"序列切片 (10-20bp): {sliced_seq}")到这里,我们也就讲完了《Python基因组处理,Biopython入门指南》的内容了。个人认为,基础知识的学习和巩固,是为了更好的将其运用到项目中,欢迎关注golang学习网公众号,带你了解更多关于的知识点!
 Cookie、LocalStorage与IndexedDB对比解析
Cookie、LocalStorage与IndexedDB对比解析
- 上一篇
- Cookie、LocalStorage与IndexedDB对比解析

- 下一篇
- Pythonsuper()使用详解与实战教程
-
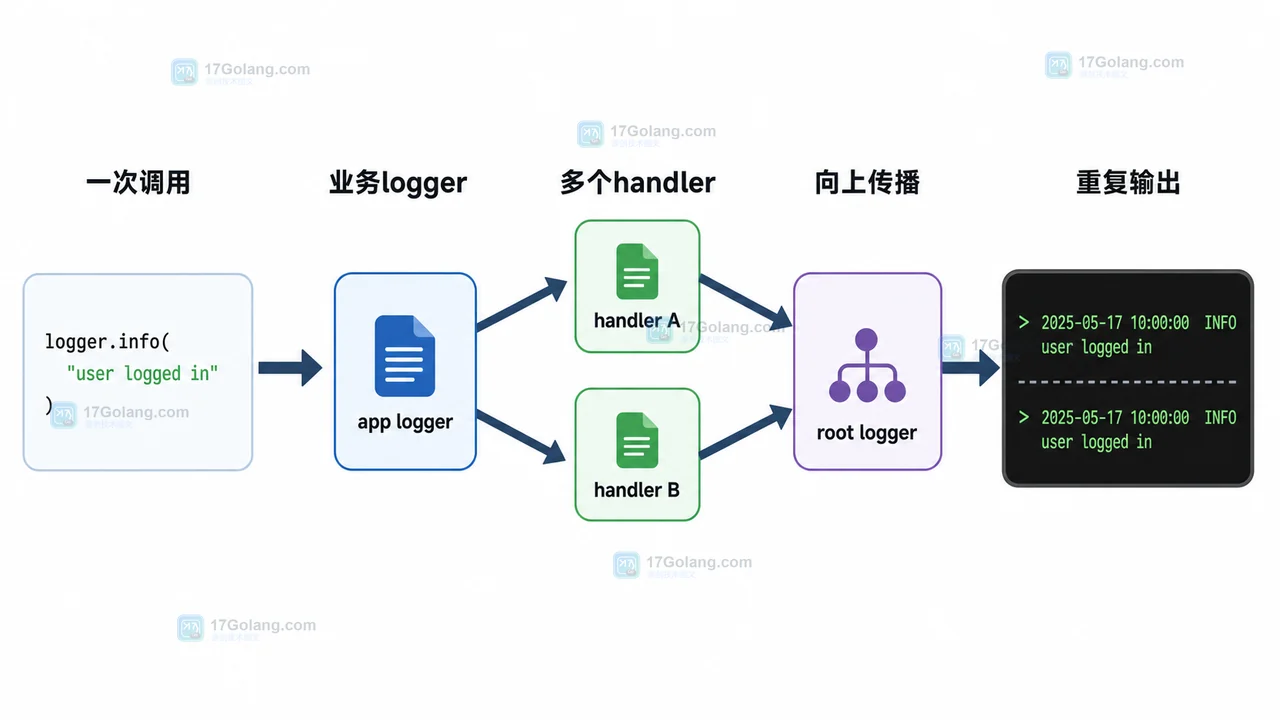
- 文章 · python教程 | 1星期前 | logging · Python教程 · 后端开发 · 日志排查 · Python logging 日志重复 propagate addHandler basicConfig
- Python logging 日志重复打印排查:为什么一条记录输出了两遍
- 324浏览 收藏
-

- 文章 · python教程 | 2星期前 | 默认值 · python · 数据建模 · dataclass · default_factory · field · Python 数据类 Field 可变默认值 dataclass default_factory
- Python dataclass 默认值完整工作流:从可变默认值到 default_factory
- 228浏览 收藏

-

- 前端进阶之JavaScript设计模式
- 设计模式是开发人员在软件开发过程中面临一般问题时的解决方案,代表了最佳的实践。本课程的主打内容包括JS常见设计模式以及具体应用场景,打造一站式知识长龙服务,适合有JS基础的同学学习。
- 543次学习
-

- GO语言核心编程课程
- 本课程采用真实案例,全面具体可落地,从理论到实践,一步一步将GO核心编程技术、编程思想、底层实现融会贯通,使学习者贴近时代脉搏,做IT互联网时代的弄潮儿。
- 516次学习
-

- 简单聊聊mysql8与网络通信
- 如有问题加微信:Le-studyg;在课程中,我们将首先介绍MySQL8的新特性,包括性能优化、安全增强、新数据类型等,帮助学生快速熟悉MySQL8的最新功能。接着,我们将深入解析MySQL的网络通信机制,包括协议、连接管理、数据传输等,让
- 500次学习
-

- JavaScript正则表达式基础与实战
- 在任何一门编程语言中,正则表达式,都是一项重要的知识,它提供了高效的字符串匹配与捕获机制,可以极大的简化程序设计。
- 487次学习
-

- 从零制作响应式网站—Grid布局
- 本系列教程将展示从零制作一个假想的网络科技公司官网,分为导航,轮播,关于我们,成功案例,服务流程,团队介绍,数据部分,公司动态,底部信息等内容区块。网站整体采用CSSGrid布局,支持响应式,有流畅过渡和展现动画。
- 485次学习
-

- ljg-skills
- ljg-skills 是李继刚开源的 AI 技能与提示词集合,面向大模型使用者整理了一批可复用的 prompt、角色设定和任务技能模板,适合用于学习提示词设计、搭建个人 AI 工作流和沉淀团队常用智能体能力。
- 3761次使用
-

- MELO音乐
- MELO音乐是一站式AI视频与音乐制作助手,对标suno, udio的高品质体验。提供伴奏生成、原创写词、无损导出、哼唱识曲、混音变声等全套音频与短视频编辑工具。无论是流行Kpop、电音说唱、民谣古风、摇滚儿歌还是商用轻音乐,MELO为你免费谱曲,轻松做同款!
- 3474次使用
-

- UniScribe
- UniScribe 是一款 AI 音视频转文字与内容整理工具,支持上传音频、视频文件或粘贴 YouTube 链接,自动生成转写文本、摘要、思维导图和关键问题,并支持多格式导出,适合会议记录、课程学习、访谈整理和内容创作复盘。
- 3444次使用
-

- 剧云
- 剧云是专业中文剧本创作平台,安全稳定运行十余年,集成AI编剧、剧本医生审核、人物小传、剧情关系图、大纲编写、多人协作、Word导入导出、版权管控功能,数据安全防护,轻松高效创作剧本。
- 3628次使用
-

- 万象有声
- 万象有声,一个专为有声创作者打造的新一代智能有声内容创作平台。平台提供专业的智能拆章、智能画本编辑、AI配音、AI生成音效、后期制作、智能对轨、智能审听等有声创作全流程工具,可以帮助创作者高效、低成本创作出引人入胜的有声作品。立即体验,让有声书制作更简单!
- 3601次使用
-
- Python监控网页状态:requests异常处理实战
- 2026-05-29 501浏览
-
- TensorFlow模型部署为API的TF Serving方法
- 2026-05-26 501浏览
-
- Python字符串编码转换:encode与decode详解
- 2026-05-16 501浏览
-
- TensorFlow裁剪无用算子方法详解
- 2026-05-15 501浏览
-
- httpx 如何设置代理认证(Proxy-Authorization)
- 2026-05-05 501浏览


