Python处理VCF文件入门指南
golang学习网今天将给大家带来《Python处理VCF文件教程》,感兴趣的朋友请继续看下去吧!以下内容将会涉及到等等知识点,如果你是正在学习文章或者已经是大佬级别了,都非常欢迎也希望大家都能给我建议评论哈~希望能帮助到大家!
Python处理VCF文件的核心库是PyVCF,它提供直观的接口解析VCF元信息、表头和变异记录。1. 安装PyVCF:使用pip install PyVCF;2. 读取VCF文件:通过vcf.Reader对象逐行解析;3. 提取核心字段:如CHROM、POS、REF、ALT、QUAL、FILTER、INFO及样本基因型;4. 过滤并写入新文件:根据QUAL和FILTER条件筛选变异并用vcf.Writer保存。此外,面对大规模VCF数据时可选用cyvcf2或pysam以提升性能。VCF结构包括元信息行(##开头)、表头行(#开头)和数据行,分别定义格式规范、列名顺序和具体变异信息。常见挑战包括格式不规范、内存占用高、INFO/FORAMT字段复杂性及缺失值问题,应对策略包括验证文件、流式处理、动态解析字段和空值检查。掌握PyVCF基础并结合高性能库是高效处理VCF的关键路径。

Python在生物信息学领域,尤其是处理VCF(Variant Call Format)文件时,简直是神器。它提供了一系列强大且灵活的库,让你能轻松地读取、解析、过滤甚至修改这些包含了遗传变异信息的文本文件。对我个人而言,无论是做科研项目还是日常数据分析,Python都是我处理VCF的首选工具,因为它不仅高效,而且社区支持非常活跃,遇到问题总能找到解决方案。

解决方案
要使用Python操作VCF文件,最常用且功能强大的库之一是PyVCF。它提供了一个直观的接口来解析VCF的各个部分,包括元信息、表头以及每一行的变异记录。
首先,你需要安装它:
pip install PyVCF
接着,你可以这样读取并处理一个VCF文件:

import vcf
# 假设你有一个名为 'example.vcf' 的VCF文件
vcf_file_path = 'example.vcf'
try:
# 创建一个VCF Reader对象
# 如果文件是gzipped的,PyVCF会自动处理
vcf_reader = vcf.Reader(open(vcf_file_path, 'r'))
# 打印VCF文件的元信息 (如文件格式版本、INFO/FORMAT定义等)
print("VCF文件元信息:")
for key, value in vcf_reader.metadata.items():
print(f" {key}: {value}")
print("\n")
# 遍历每一条变异记录
print("前5条变异记录示例:")
record_count = 0
for record in vcf_reader:
if record_count >= 5:
break
# 访问核心字段
chrom = record.CHROM
pos = record.POS
ref = record.REF
alts = [str(a) for a in record.ALT] # ALT可能是一个列表
qual = record.QUAL
filters = record.FILTER # 过滤状态,可能是None或列表
info = record.INFO # INFO字段是一个字典
print(f" CHROM: {chrom}, POS: {pos}, REF: {ref}, ALT: {alts}, QUAL: {qual}")
print(f" FILTER: {filters}")
print(f" INFO (部分): {info.get('DP', 'N/A')} (深度), {info.get('AF', 'N/A')} (等位基因频率)")
# 访问样本基因型信息
for sample in record.samples:
sample_name = sample.sample
gt = sample['GT'] # 基因型,如'0/1', '1/1'
dp = sample['DP'] # 样本深度
ad = sample['AD'] # 等位基因深度 (REF, ALT)
print(f" 样本 {sample_name}: GT={gt}, DP={dp}, AD={ad}")
print("-" * 30)
record_count += 1
# 示例:过滤特定质量的变异并写入新文件
output_vcf_path = 'filtered_variants.vcf'
with open(output_vcf_path, 'w') as output_handle:
vcf_writer = vcf.Writer(output_handle, vcf_reader) # 传入reader的header信息
# 重置reader以重新遍历
vcf_reader = vcf.Reader(open(vcf_file_path, 'r'))
filtered_count = 0
for record in vcf_reader:
# 假设我们只保留质量大于30且没有被FILTER标记的变异
if record.QUAL is not None and record.QUAL > 30 and (record.FILTER is None or 'PASS' in record.FILTER):
vcf_writer.write_record(record)
filtered_count += 1
print(f"\n成功过滤并写入 {filtered_count} 条变异到 {output_vcf_path}")
except FileNotFoundError:
print(f"错误: 文件 '{vcf_file_path}' 未找到。请确保文件路径正确。")
except Exception as e:
print(f"处理VCF文件时发生错误: {e}")
这个代码片段展示了VCF文件读写和基本过滤操作。你会发现,PyVCF将VCF文件中的每一行数据抽象为一个Record对象,其属性直接对应VCF规范中的字段,非常直观。
为什么VCF文件在生物信息学中如此重要,以及其基本结构是什么?
VCF文件(Variant Call Format)在生物信息学领域的重要性不言而喻,它几乎是所有高通量测序数据变异检测的“通用语言”。想象一下,我们从一个人或一个物种的DNA中检测到了成千上万甚至数百万个与参考基因组不同的地方,VCF就是用来标准地记录这些差异(变异)的格式。它的重要性在于:它提供了一个结构化、可机器解析的方式来存储基因组变异信息,包括单核苷酸多态性(SNPs)、插入缺失(Indels)、结构变异(SVs)等。如果没有VCF,大家各自为政,数据共享和下游分析将寸步难行。
从结构上看,一个VCF文件可以大致分为三个部分:
元信息行 (Meta-information Lines):以
##开头。这些行提供了关于VCF文件本身的描述信息,比如VCF格式的版本号 (##fileformat)、对INFO字段的详细解释 (##INFO)、对FORMAT字段的定义 (##FORMAT)、对FILTER字段的描述 (##FILTER),以及其他任何与变异调用过程相关的元数据。这些信息对于理解VCF文件中数据的含义至关重要。表头行 (Header Line):以单个
#开头。这一行定义了后续数据列的名称和顺序。标准列包括:#CHROM: 染色体名称。POS: 变异在染色体上的起始位置(1-based)。ID: 变异的唯一标识符(如rs号),如果没有则为.。REF: 参考基因组在该位置的碱基或序列。ALT: 变异等位基因的碱基或序列。QUAL: 变异的质量值,越高代表越可靠。FILTER: 过滤状态,表示变异是否通过了质量过滤。PASS表示通过,否则会列出失败的原因。INFO: 一个包含多个键值对的字段,用于存储变异的额外信息,如等位基因频率(AF)、深度(DP)等。FORMAT: 定义了后续样本列中基因型信息的格式。- 随后的列是样本列,每一列代表一个样本,其中包含该样本在该变异位点上的基因型信息,其具体内容由
FORMAT列定义。
数据行 (Data Lines):不以
#开头的行。每一行代表一个独立的变异事件,按照表头行定义的顺序填充相应的数据。这些行是VCF文件的核心,承载了所有具体的变异位点信息及其在各个样本中的状态。
理解这些结构对于正确解析和利用VCF数据至关重要,特别是INFO和FORMAT字段,它们是VCF灵活性的关键,也是初学者常常感到困惑的地方。
除了PyVCF,还有哪些Python库可以高效处理大规模VCF数据?
虽然PyVCF对于大多数常规VCF操作来说已经足够好用,但在处理大规模VCF文件时,尤其是在需要极致性能的场景下,它的表现可能就不那么尽如人意了。这时,我通常会考虑其他一些专门为性能优化的Python库:
cyvcf2: 这是我个人在处理TB级别VCF数据时首选的库。cyvcf2是基于Cython编写的,它直接绑定了C语言的htslib库(这是SAMtools、BCFtools等生物信息学工具的基础库),因此在读取速度和内存效率上都有显著优势。它能非常快速地迭代VCF记录,并且对Gzipped VCF文件(.vcf.gz)的支持也非常好。如果你面临的VCF文件动辄几GB甚至几十GB,cyvcf2绝对值得一试。它的API设计也比较简洁,用起来非常顺手,尤其是在需要快速过滤或提取特定字段时。# 安装 # pip install cyvcf2 # 简单使用示例 from cyvcf2 import VCF vcf_file = 'large_example.vcf.gz' vcf_reader = VCF(vcf_file) print(f"使用cyvcf2读取前5条记录:") record_count = 0 for variant in vcf_reader: if record_count >= 5: break # 访问字段方式略有不同,但同样直观 print(f" CHROM: {variant.CHROM}, POS: {variant.POS}, REF: {variant.REF}, ALT: {variant.ALT}") # 获取INFO字段 print(f" INFO: DP={variant.INFO.get('DP')}, AF={variant.INFO.get('AF')}") # 获取样本基因型 # variant.genotypes 是一个NumPy数组,包含GT, AD, DP等 # 比如,第一个样本的基因型 # print(f" Sample 0 GT: {variant.gt_types[0]}, AD: {variant.gt_depths[0]}, DP: {variant.gt_ref_depths[0]}, {variant.gt_alt_depths[0]}") record_count += 1你会发现,
cyvcf2的接口设计考虑了性能,例如variant.gt_types直接返回整数编码的基因型,variant.gt_depths返回深度数组,这在处理大量样本时非常高效。pysam:pysam是一个Python封装,它同样绑定了htslib库,因此它不仅能处理VCF/BCF文件,还能处理SAM/BAM/CRAM等序列比对文件。如果你在一个项目中需要同时操作多种生物信息学文件格式,那么pysam是一个非常全面的选择。它在VCF处理方面也提供了高性能的读写能力,并且支持BCF(Binary Call Format),这是一种VCF的二进制压缩格式,能进一步提升读写效率和存储空间。虽然它的API可能不如PyVCF或cyvcf2那样专注于VCF,但其多功能性使其成为许多生物信息学管道中不可或缺的工具。
选择哪个库,很大程度上取决于你的具体需求:如果只是偶尔处理一些中小型VCF文件,PyVCF的易用性是优势;如果经常面对大规模数据集,并且对处理速度有较高要求,那么cyvcf2或pysam(特别是cyvcf2)会是更明智的选择。在我看来,掌握PyVCF的基础,再根据实际项目需要深入学习cyvcf2,是比较理想的学习路径。
在处理VCF文件时,常见的挑战和错误有哪些,如何避免?
处理VCF文件,特别是那些来自不同来源或不同变异调用工具生成的VCF,往往会遇到一些让人头疼的挑战和错误。这些问题可能会导致你的脚本崩溃,或者更隐蔽地,产生错误的结果。作为过来人,我总结了一些常见的“坑”和我的应对策略:
VCF文件格式不规范或版本不一致:
- 挑战:VCF规范虽然存在,但实际生成的文件可能存在细微的偏差,例如缺少必要的元信息行,或者
INFO/FORMAT字段的定义与实际数据不符。不同版本的VCF规范(如VCFv4.1 vs VCFv4.2)在某些细节上也有差异。 - 错误表现:解析库报错,或者某个字段解析出来是
None或不符合预期。 - 避免:
- 验证:在处理前,使用
vcftools --vcf-validator或bcftools check等工具对VCF文件进行初步验证。这能帮你发现很多低级格式错误。 - 宽容解析:如果条件允许,你的代码应该对一些不那么严格的格式保持一定的宽容度。例如,当某个
INFO字段不存在时,不要直接报错,而是给予默认值或跳过。 - 理解元信息:解析
INFO和FORMAT字段时,务必先读取并理解VCF头部的元信息定义。这些定义告诉你每个键的类型(整数、浮点数、字符串)和数量(单个值、数组、与ALT等位基因数量相关等),这对于正确解析至关重要。
- 验证:在处理前,使用
- 挑战:VCF规范虽然存在,但实际生成的文件可能存在细微的偏差,例如缺少必要的元信息行,或者
处理大型VCF文件时的内存和性能问题:
- 挑战:全基因组测序的VCF文件可能非常庞大,几十GB甚至上百GB是常态。一次性将整个文件加载到内存中几乎是不可能的,会导致内存溢出(OOM)。
- 错误表现:脚本运行缓慢,内存占用飙升,最终崩溃。
- 避免:
- 流式处理:使用像
PyVCF、cyvcf2或pysam这样的库,它们都支持流式读取,即每次只加载一条或一小批记录到内存中进行处理。这是处理大文件的基本原则。 - 选择高性能库:如前所述,对于性能敏感的任务,优先使用
cyvcf2或pysam,它们底层基于C语言库,效率更高。 - 分块处理:如果需要对文件进行多次遍历或复杂操作,考虑将大文件分割成更小的块进行处理,或者利用数据库(如Tabix索引的VCF文件)进行随机访问。
- BCF格式:如果你的工具链支持,将VCF转换为BCF(二进制VCF)格式。BCF文件更紧凑,读取速度也更快。
- 流式处理:使用像
INFO和FORMAT字段的复杂性与多样性:
- 挑战:
INFO和FORMAT字段是VCF灵活性的来源,但也是复杂性的根源。它们可以包含各种自定义的键值对,其含义、类型和数量都可能因变异调用工具、分析流程甚至项目而异。比如,一个INFO字段AF(等位基因频率)可能是一个浮点数,也可能是一个与ALT等位基因数量对应的浮点数列表。 - 错误表现:解析出的数据类型不正确,或者无法正确关联到对应的等位基因。
- 避免:
- 查阅文档:总是查阅生成VCF文件的工具的文档,了解其输出的
INFO和FORMAT字段的具体含义和类型。 - 动态解析:不要硬编码对特定
INFO/FORMAT字段的期望。利用库提供的API(如record.INFO.get('KEY'))来安全地访问,并根据元信息中的定义来处理其类型和数量。 - 类型转换:从VCF解析出的某些值可能是字符串,如果需要进行数值计算,务必进行显式的类型转换(
int(),float()),并处理可能出现的转换错误。
- 查阅文档:总是查阅生成VCF文件的工具的文档,了解其输出的
- 挑战:
处理缺失值和空值:
- 挑战:VCF文件中经常会出现缺失值(用
.表示),或者某些字段根本不存在。例如,QUAL字段可能为.,FILTER字段可能为None。 - 错误表现:在对这些值进行操作时(如
record.QUAL > 30),如果record.QUAL是None或.,会导致类型错误。 - 避免:
- 空值检查:在对任何可能缺失的字段进行操作前,始终进行空值检查(
if record.QUAL is not None:)。 - 默认值:为缺失值设置合理的默认值,例如,如果质量值缺失,可以将其视为0或一个非常小的值。
- 空值检查:在对任何可能缺失的字段进行操作前,始终进行空值检查(
- 挑战:VCF文件中经常会出现缺失值(用
处理VCF文件是一个迭代和学习的过程。一开始可能会遇到很多挫折,但随着经验的积累和对VCF规范的深入理解,你会发现它其实是一个设计得相当精妙且强大的数据格式。多动手,多尝试,是最好的学习方式。
以上就是《Python处理VCF文件入门指南》的详细内容,更多关于的资料请关注golang学习网公众号!
 JavaJDBC批量操作怎么用,优势有哪些?
JavaJDBC批量操作怎么用,优势有哪些?
- 上一篇
- JavaJDBC批量操作怎么用,优势有哪些?

- 下一篇
- Python音频处理:pydub入门教程详解
-
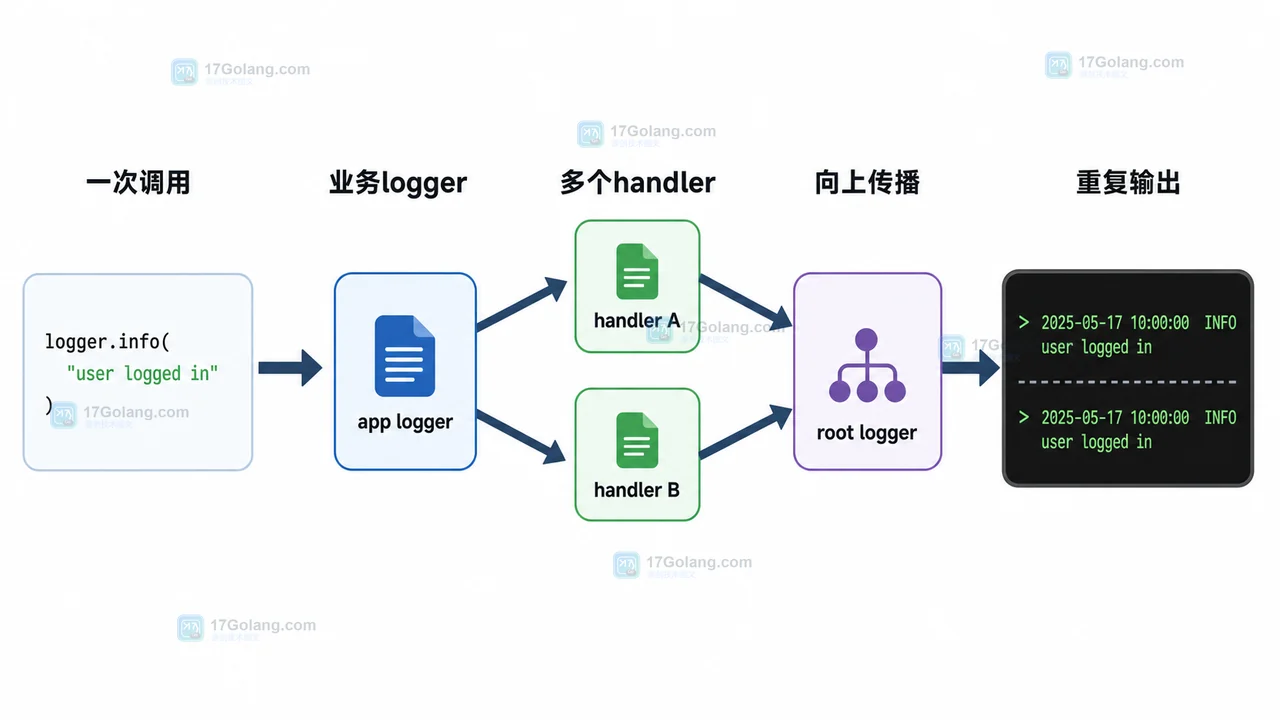
- 文章 · python教程 | 3天前 | logging · Python教程 · 后端开发 · 日志排查 · Python logging 日志重复 propagate addHandler basicConfig
- Python logging 日志重复打印排查:为什么一条记录输出了两遍
- 324浏览 收藏
-

- 文章 · python教程 | 2星期前 | 默认值 · python · 数据建模 · dataclass · default_factory · field · Python 数据类 Field 可变默认值 dataclass default_factory
- Python dataclass 默认值完整工作流:从可变默认值到 default_factory
- 228浏览 收藏

-

- 前端进阶之JavaScript设计模式
- 设计模式是开发人员在软件开发过程中面临一般问题时的解决方案,代表了最佳的实践。本课程的主打内容包括JS常见设计模式以及具体应用场景,打造一站式知识长龙服务,适合有JS基础的同学学习。
- 543次学习
-

- GO语言核心编程课程
- 本课程采用真实案例,全面具体可落地,从理论到实践,一步一步将GO核心编程技术、编程思想、底层实现融会贯通,使学习者贴近时代脉搏,做IT互联网时代的弄潮儿。
- 516次学习
-

- 简单聊聊mysql8与网络通信
- 如有问题加微信:Le-studyg;在课程中,我们将首先介绍MySQL8的新特性,包括性能优化、安全增强、新数据类型等,帮助学生快速熟悉MySQL8的最新功能。接着,我们将深入解析MySQL的网络通信机制,包括协议、连接管理、数据传输等,让
- 500次学习
-

- JavaScript正则表达式基础与实战
- 在任何一门编程语言中,正则表达式,都是一项重要的知识,它提供了高效的字符串匹配与捕获机制,可以极大的简化程序设计。
- 487次学习
-

- 从零制作响应式网站—Grid布局
- 本系列教程将展示从零制作一个假想的网络科技公司官网,分为导航,轮播,关于我们,成功案例,服务流程,团队介绍,数据部分,公司动态,底部信息等内容区块。网站整体采用CSSGrid布局,支持响应式,有流畅过渡和展现动画。
- 485次学习
-

- ljg-skills
- ljg-skills 是李继刚开源的 AI 技能与提示词集合,面向大模型使用者整理了一批可复用的 prompt、角色设定和任务技能模板,适合用于学习提示词设计、搭建个人 AI 工作流和沉淀团队常用智能体能力。
- 3169次使用
-

- MELO音乐
- MELO音乐是一站式AI视频与音乐制作助手,对标suno, udio的高品质体验。提供伴奏生成、原创写词、无损导出、哼唱识曲、混音变声等全套音频与短视频编辑工具。无论是流行Kpop、电音说唱、民谣古风、摇滚儿歌还是商用轻音乐,MELO为你免费谱曲,轻松做同款!
- 2923次使用
-

- UniScribe
- UniScribe 是一款 AI 音视频转文字与内容整理工具,支持上传音频、视频文件或粘贴 YouTube 链接,自动生成转写文本、摘要、思维导图和关键问题,并支持多格式导出,适合会议记录、课程学习、访谈整理和内容创作复盘。
- 2881次使用
-

- 剧云
- 剧云是专业中文剧本创作平台,安全稳定运行十余年,集成AI编剧、剧本医生审核、人物小传、剧情关系图、大纲编写、多人协作、Word导入导出、版权管控功能,数据安全防护,轻松高效创作剧本。
- 3087次使用
-

- 万象有声
- 万象有声,一个专为有声创作者打造的新一代智能有声内容创作平台。平台提供专业的智能拆章、智能画本编辑、AI配音、AI生成音效、后期制作、智能对轨、智能审听等有声创作全流程工具,可以帮助创作者高效、低成本创作出引人入胜的有声作品。立即体验,让有声书制作更简单!
- 3042次使用
-
- Python监控网页状态:requests异常处理实战
- 2026-05-29 501浏览
-
- TensorFlow模型部署为API的TF Serving方法
- 2026-05-26 501浏览
-
- Python字符串编码转换:encode与decode详解
- 2026-05-16 501浏览
-
- TensorFlow裁剪无用算子方法详解
- 2026-05-15 501浏览
-
- httpx 如何设置代理认证(Proxy-Authorization)
- 2026-05-05 501浏览


