BioJava序列分析教程:Java处理生物信息
目前golang学习网上已经有很多关于文章的文章了,自己在初次阅读这些文章中,也见识到了很多学习思路;那么本文《Java处理生物信息,BioJava序列分析教程》,也希望能帮助到大家,如果阅读完后真的对你学习文章有帮助,欢迎动动手指,评论留言并分享~
BioJava在序列处理中的核心优势包括跨平台性与强类型保障代码健壮性、提供全面的功能模块支持多种生物信息学任务、以及依托Java生态在大型系统集成和性能优化上的成熟支持。其挑战则体现在API学习曲线较陡、社区活跃度相对较低导致新功能迭代缓慢、以及特定高性能需求场景下可能不如C/C++实现高效。使用BioJava进行DNA/RNA常见操作的流程为:1. 创建或加载序列,可通过字符串直接构建或从FASTA等文件读取;2. 执行基本操作如获取长度、反向互补、转录RNA、翻译蛋白质、提取子序列;3. 实现高级分析如计算GC含量等。

Java在生物信息学领域,特别是序列分析方面,确实能发挥出相当大的作用。虽然Python因其简洁和丰富的库生态在生物信息学中占据主导地位,但Java凭借其强大的类型系统、JVM的跨平台能力以及在大型项目中的稳定性,尤其在处理大规模数据和构建复杂应用时,仍然是一个非常可靠且高效的选择。对于序列分析,BioJava库无疑是Java生态系统中的核心利器,它提供了一整套API来处理各种生物序列数据和算法。

解决方案
要使用Java处理生物信息,特别是进行序列分析,核心在于有效利用BioJava库。这个库封装了生物信息学中常见的概念和算法,例如序列(DNA、RNA、蛋白质)、字母表(Alphabet)、序列操作(反向互补、翻译)、文件解析(FASTA、GenBank)以及序列比对等。

一个典型的BioJava工作流程会涉及:
- 引入BioJava依赖: 通常通过Maven或Gradle将BioJava的核心模块添加到项目中。
- 加载或创建序列: 从文件(如FASTA、GenBank)中读取序列,或者直接在代码中构建序列对象。
- 执行序列操作: 利用BioJava提供的工具类(如
DNATools,RNATools,AAATools)进行各种操作,例如计算GC含量、获取反向互补序列、转录或翻译。 - 进行更复杂的分析: 如序列比对、特征提取或基于序列的模式匹配。
以下是一个简单的BioJava代码片段,展示如何创建一个DNA序列并获取其反向互补序列:
import org.biojava.nbio.core.sequence.DNASequence;
import org.biojava.nbio.core.sequence.compound.AmbiguityDNACompoundSet;
import org.biojava.nbio.core.sequence.template.Sequence;
import org.biojava.nbio.core.sequence.transcription.DNATranslator;
import org.biojava.nbio.core.sequence.io.FastaReaderHelper;
import java.io.File;
import java.io.FileInputStream;
import java.util.LinkedHashMap;
public class BioJavaSequenceExample {
public static void main(String[] args) {
// 1. 创建一个DNA序列
try {
DNASequence dnaSeq = new DNASequence("ATGCGTACGTAGCTAGCTAG");
System.out.println("原始DNA序列: " + dnaSeq.getSequenceAsString());
// 2. 获取反向互补序列
DNASequence reverseComplementSeq = dnaSeq.get = dnaSeq.get = dnaSeq.get = dnaSeq.get = dnaSeq.getReverseComplement();
System.out.println("反向互补序列: " + reverseComplementSeq.getSequenceAsString());
// 3. 转录为RNA序列 (虽然是DNASequence对象,但可以执行转录操作)
Sequence rnaSeq = DNATranslator.transcribe(dnaSeq);
System.out.println("转录后的RNA序列: " + rnaSeq.getSequenceAsString());
// 4. 尝试从FASTA文件读取序列 (假设存在一个test.fasta文件)
// 这是一个概念性的示例,实际使用需要文件存在
// File fastaFile = new File("test.fasta");
// if (fastaFile.exists()) {
// LinkedHashMap dnaSequences = FastaReaderHelper.readFastaDNASequence(fastaFile);
// for (DNASequence seq : dnaSequences.values()) {
// System.out.println("从FASTA读取的序列: " + seq.getSequenceAsString());
// break; // 示例只读取第一个
// }
// } else {
// System.out.println("test.fasta 文件不存在,跳过文件读取示例。");
// System.out.println("可以创建一个包含 '>seq1\\nATGC' 的test.fasta文件来测试。");
// }
} catch (Exception e) {
e.printStackTrace();
}
}
} BioJava在序列处理中的核心优势与挑战是什么?
在我看来,BioJava在序列处理方面确实有一些独特的优势,但也伴随着一些不容忽视的挑战。
优势方面: 首先,作为Java生态的一部分,BioJava继承了Java语言的跨平台性和强类型特性。这意味着你编写的代码可以在任何支持JVM的环境中运行,并且编译时就能发现很多类型相关的错误,这对于构建大型、复杂的生物信息学系统来说,无疑增加了代码的健壮性和可维护性。我个人很喜欢Java的这种“严格”,它能帮助团队在项目初期就避免很多潜在的问题。
其次,BioJava提供了相当全面的功能模块。从基本的序列操作、文件解析(FASTA, GenBank, PDB等),到更高级的序列比对、结构分析,甚至是对生物本体论(Ontology)的支持,它几乎涵盖了生物信息学中常用的各个方面。这意味着开发者在一个框架内就能完成大部分工作,减少了集成不同工具的麻烦。
再者,Java在企业级应用和高性能计算方面有着深厚的积累。如果你的生物信息学分析需要处理PB级别的数据,或者需要与现有的企业级系统(如数据库、消息队列)深度集成,Java的生态系统和性能优化工具链会比一些脚本语言更成熟。JVM的垃圾回收机制和JIT编译器在处理长时间运行的、内存密集型任务时,也能提供不错的性能保障。
挑战方面: 然而,BioJava也有其“硬币的另一面”。 最明显的挑战可能就是学习曲线相对陡峭。BioJava的设计哲学偏向于面向对象和接口,这使得它的API结构比较严谨,但对于初学者来说,理解其复杂的类层次结构和各种抽象概念可能需要一些时间。相比之下,Python的Biopython则显得更加“平易近人”,很多操作一行代码就能搞定,这让很多快速原型开发更倾向于Python。
另一个挑战是社区活跃度。虽然BioJava是一个成熟且功能强大的库,但相较于Biopython或R语言的生物信息学包,其社区活跃度和新功能迭代速度可能显得略慢。这意味着当你遇到一些非常新颖或边缘化的生物信息学问题时,可能需要更多地依赖自己去实现或查找较少的现有解决方案。
最后,性能调优在特定场景下也可能成为一个挑战。尽管Java本身性能不俗,但在处理一些对计算资源极致敏感的算法(例如大规模的序列比对,尤其是需要自定义矩阵或复杂参数时),纯Java的实现可能不如C/C++编写的专业工具(如BLAST、HMMER)那样快。当然,这通常可以通过调用外部进程或使用JNI来解决,但这又增加了系统的复杂性。所以,选择Java时,你需要权衡开发效率和极致性能的需求。
如何利用BioJava进行DNA/RNA序列的常见操作?
利用BioJava进行DNA/RNA序列的常见操作,主要是通过其核心的Sequence接口及其具体实现类(如DNASequence, RNASequence)以及辅助工具类(如DNATools, RNATools)来完成的。这些工具类提供了丰富的方法,让你可以方便地处理序列数据。
1. 创建和加载序列: 你可以直接从字符串创建序列,或者从FASTA、GenBank等文件格式中加载。
从字符串创建:
import org.biojava.nbio.core.sequence.DNASequence; import org.biojava.nbio.core.sequence.RNASequence; import org.biojava.nbio.core.sequence.compound.AmbiguityDNACompoundSet; import org.biojava.nbio.core.sequence.compound.AmbiguityRNACompoundSet; // 创建DNA序列 DNASequence dnaSeq = new DNASequence("ATGCGTACGTAGCTAGCTAG"); System.out.println("DNA序列: " + dnaSeq.getSequenceAsString()); // 创建RNA序列 RNASequence rnaSeq = new RNASequence("AUGGCUACGUAGCUAGCUG"); System.out.println("RNA序列: " + rnaSeq.getSequenceAsString());从FASTA文件加载: BioJava提供了
FastaReaderHelper来简化FASTA文件的读取。import org.biojava.nbio.core.sequence.io.FastaReaderHelper; import java.io.File; import java.util.LinkedHashMap; File fastaFile = new File("path/to/your/sequences.fasta"); try { LinkedHashMap对于RNA序列,可以使用
FastaReaderHelper.readFastaRNASequence(fastaFile)。
2. 序列基本操作:
获取序列长度:
int length = dnaSeq.getLength(); System.out.println("序列长度: " + length);获取反向互补序列 (DNA): 这是DNA序列分析中非常常见的操作。
DNASequence reverseComplement = dnaSeq.getReverseComplement(); System.out.println("反向互补序列: " + reverseComplement.getSequenceAsString());转录 (DNA -> RNA): 将DNA序列转录为RNA序列。
import org.biojava.nbio.core.sequence.transcription.DNATranslator; import org.biojava.nbio.core.sequence.template.Sequence; Sequence transcribedRNA = DNATranslator.transcribe(dnaSeq); System.out.println("转录后的RNA序列: " + transcribedRNA.getSequenceAsString());翻译 (RNA -> 蛋白质): 将RNA序列翻译为蛋白质序列。需要注意,
DNATranslator也可以直接从DNA翻译,它会先进行转录。import org.biojava.nbio.core.sequence.transcription.RNATranslator; import org.biojava.nbio.core.sequence.template.Sequence; import org.biojava.nbio.core.sequence.ProteinSequence; // 如果是DNA序列,先转录再翻译 ProteinSequence proteinFromDNA = DNATranslator.translate(dnaSeq); System.out.println("从DNA翻译的蛋白质序列: " + proteinFromDNA.getSequenceAsString()); // 如果是RNA序列,直接翻译 ProteinSequence proteinFromRNA = RNATranslator.translate(rnaSeq); System.out.println("从RNA翻译的蛋白质序列: " + proteinFromRNA.getSequenceAsString());提取子序列:
// 提取从索引2(第三个碱基)到索引5(第六个碱基)的子序列 DNASequence subSeq = dnaSeq.getSubSequence(2, 5); System.out.println("子序列 (2-5): " + subSeq.getSequenceAsString());计算GC含量: BioJava没有直接的
getGCContent()方法,但你可以通过遍历序列并计数来实现。long gcCount = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().get = dnaSeq.getCompoundSet().getCompounds().stream() .filter(c -> c.equals(AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompoundSet.get=AmbiguityDNACompound
理论要掌握,实操不能落!以上关于《BioJava序列分析教程:Java处理生物信息》的详细介绍,大家都掌握了吧!如果想要继续提升自己的能力,那么就来关注golang学习网公众号吧!
 Python循环优化:numpy向量化提速技巧
Python循环优化:numpy向量化提速技巧
- 上一篇
- Python循环优化:numpy向量化提速技巧

- 下一篇
- MySQL缓存优化技巧分享
-
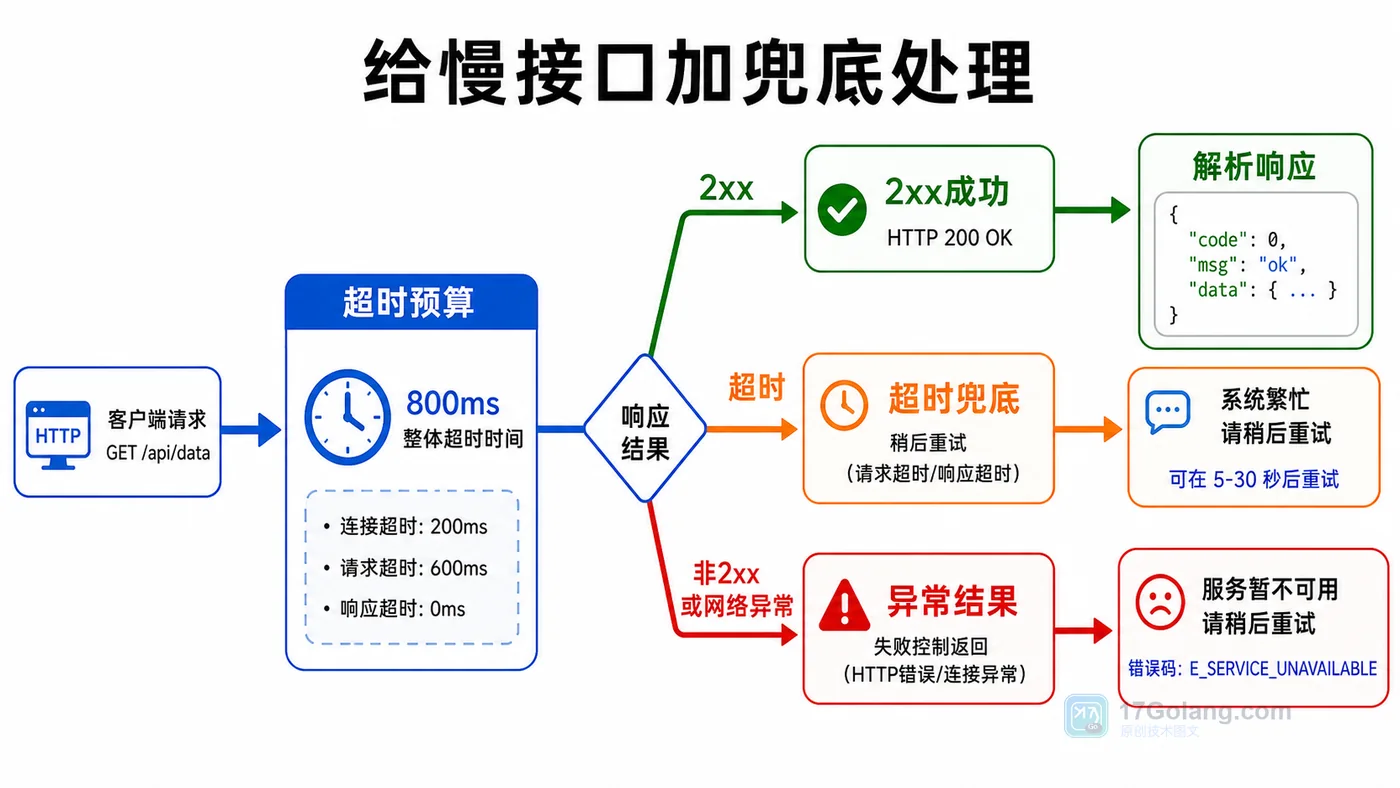
- 文章 · java教程 | 1天前 | http接口 · httpclient · Java教程 · 接口调试 · 超时处理 · java 接口调用 httpclient 超时控制 状态码 响应体
- Java HttpClient 调接口实战:超时、状态码和响应体这样处理
- 224浏览 收藏
-
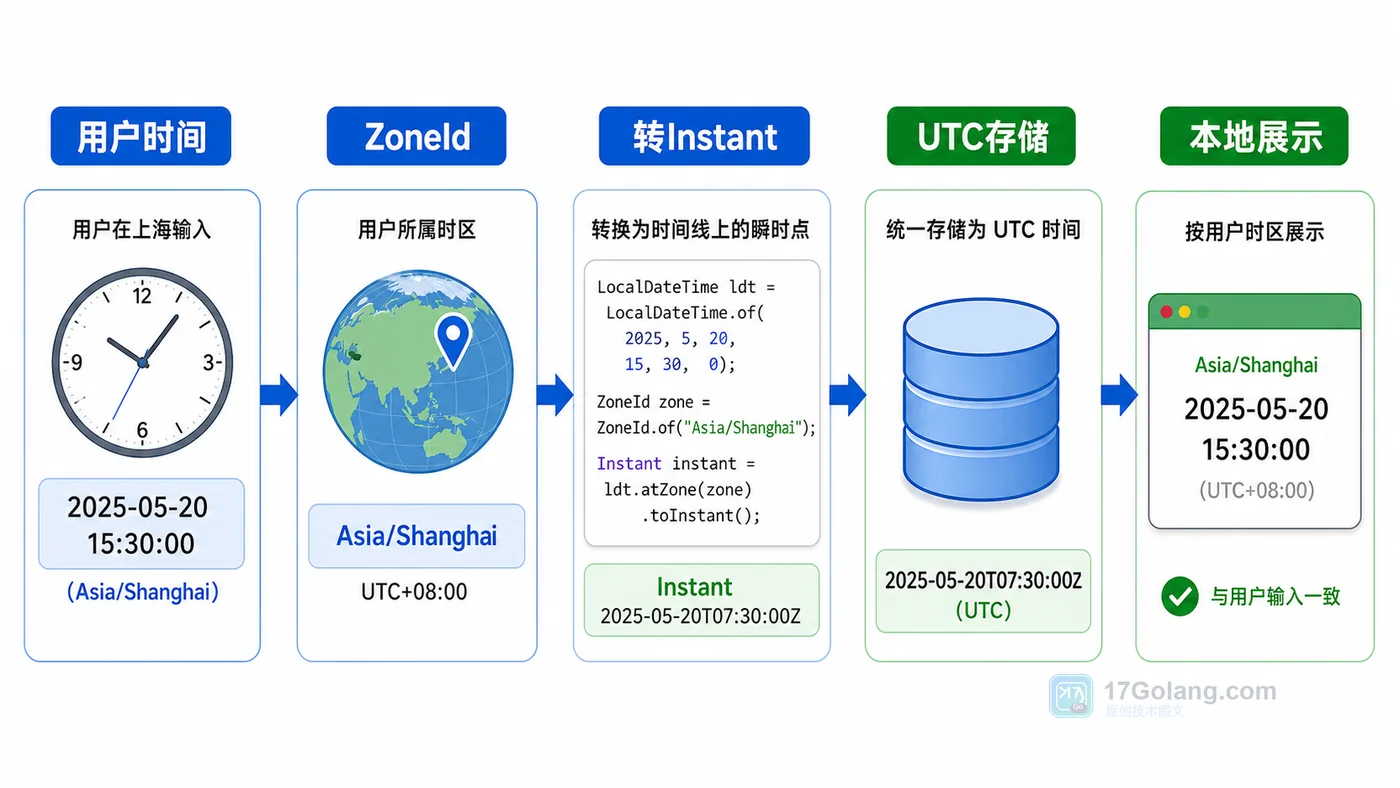
- 文章 · java教程 | 1天前 | 时间处理 · instant · Java教程 · 时区转换 · DateTimeFormatter · java DateTimeFormatter java.time 时区处理 ZoneId INSTANT
- Java 时间与时区处理实战:Instant、ZoneId 和 DateTimeFormatter 怎么配
- 461浏览 收藏
-
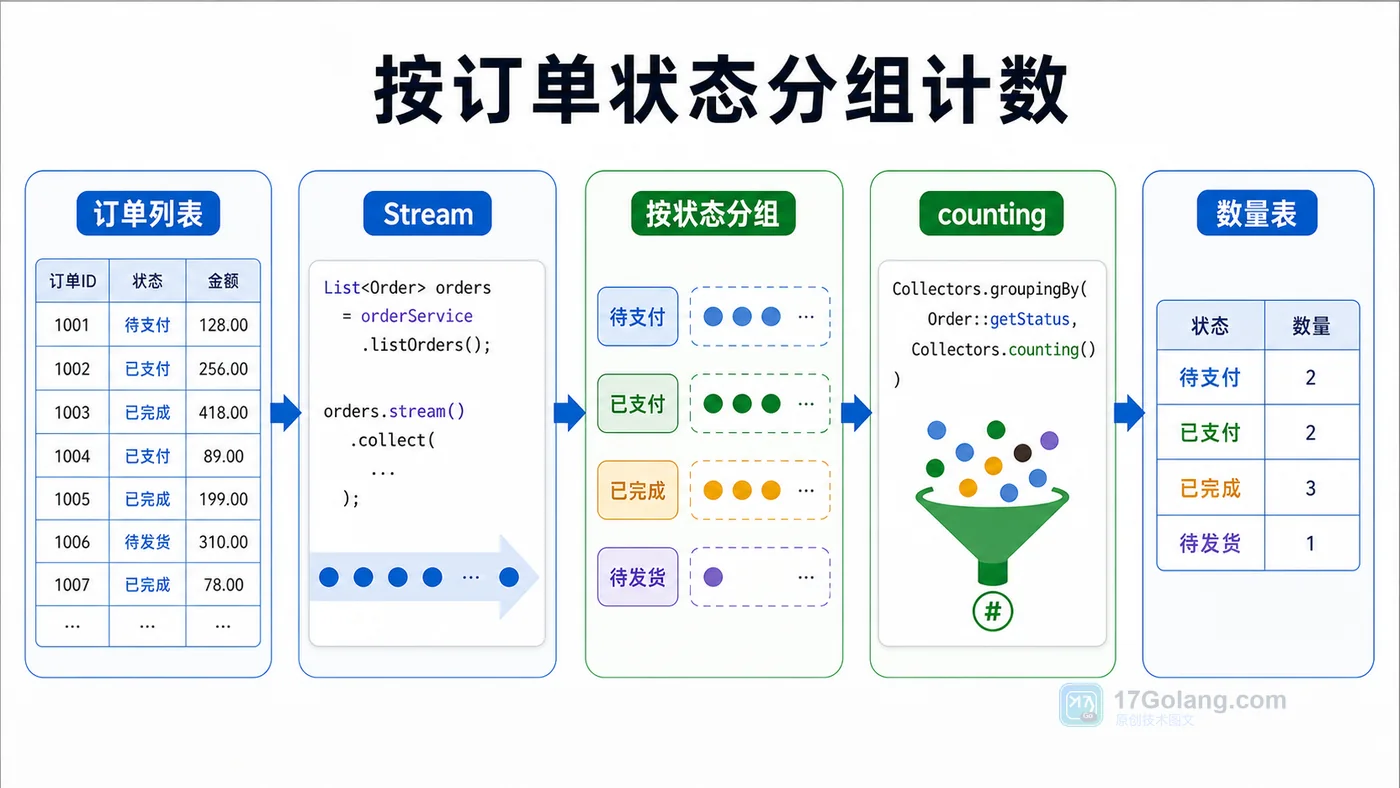
- 文章 · java教程 | 1天前 | Java · Stream · 集合统计 · 分组聚合 · Collectors · java Stream Collectors groupingBy counting summarizingInt
- Java Stream 分组统计实战:groupingBy、counting 和 summarizingInt 怎么用
- 478浏览 收藏
-
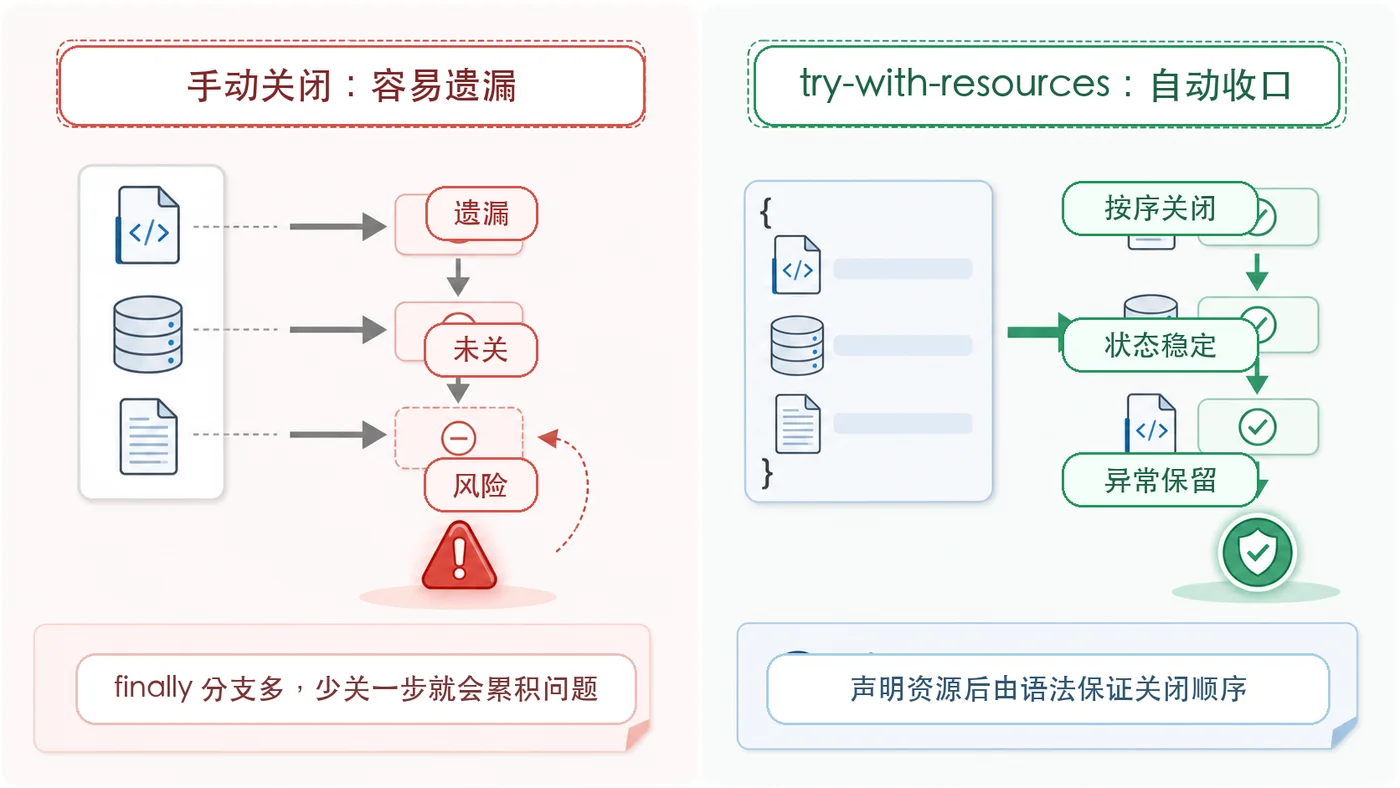
- 文章 · java教程 | 2天前 | Java · 文件读取 · 异常处理 · 资源管理 · try-with-resources · java 异常处理 try-with-resources 资源关闭 AutoCloseable 文件流
- Java try-with-resources 资源关闭实战:文件流和目录扫描这样写更稳
- 268浏览 收藏
-
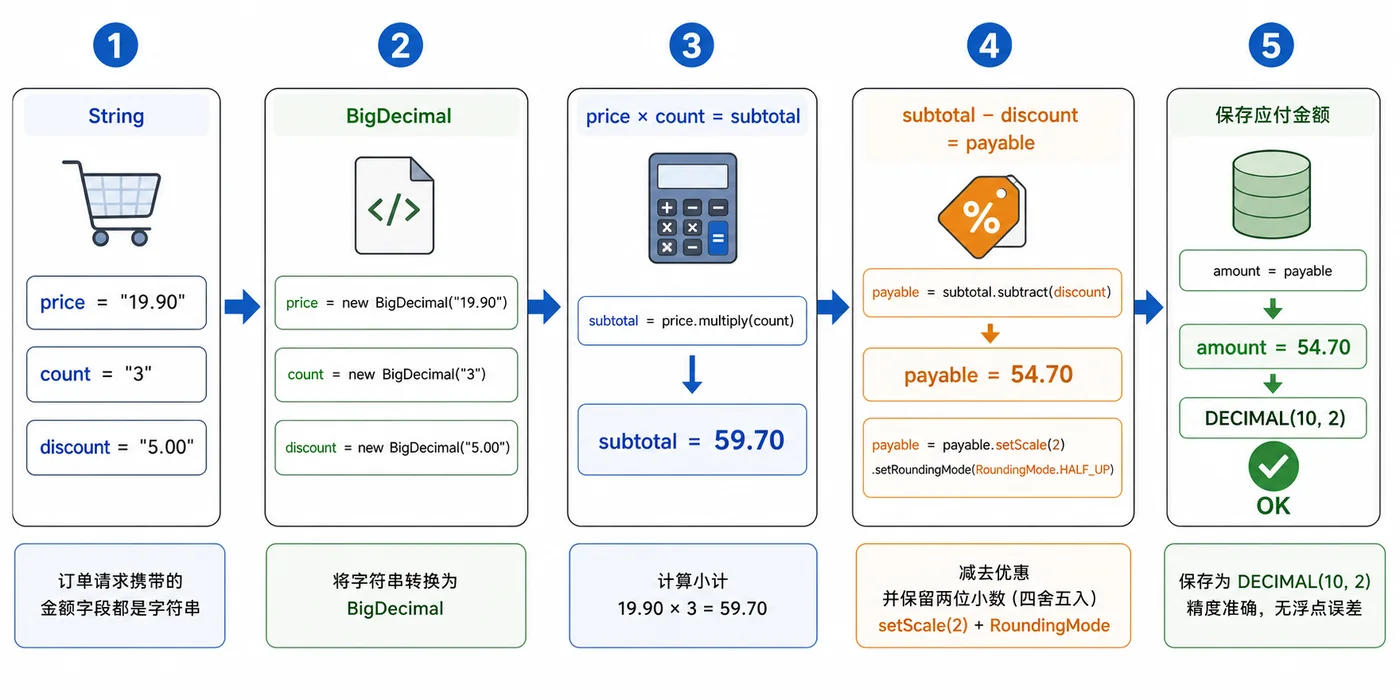
- 文章 · java教程 | 2天前 | Java教程 · 后端开发 · BigDecimal · 金额计算 · java 舍入 bigdecimal 浮点误差 金额计算 RoundingMode
- Java BigDecimal 金额计算实战:避免浮点误差和舍入问题
- 324浏览 收藏
-
- 文章 · java教程 | 2天前 | 异步编程 · Java教程 · 超时治理 · CompletableFuture · java 异步任务 超时处理 completablefuture orTimeout completeOnTimeout
- Java CompletableFuture 超时处理实战:orTimeout 和兜底结果怎么选
- 421浏览 收藏
-

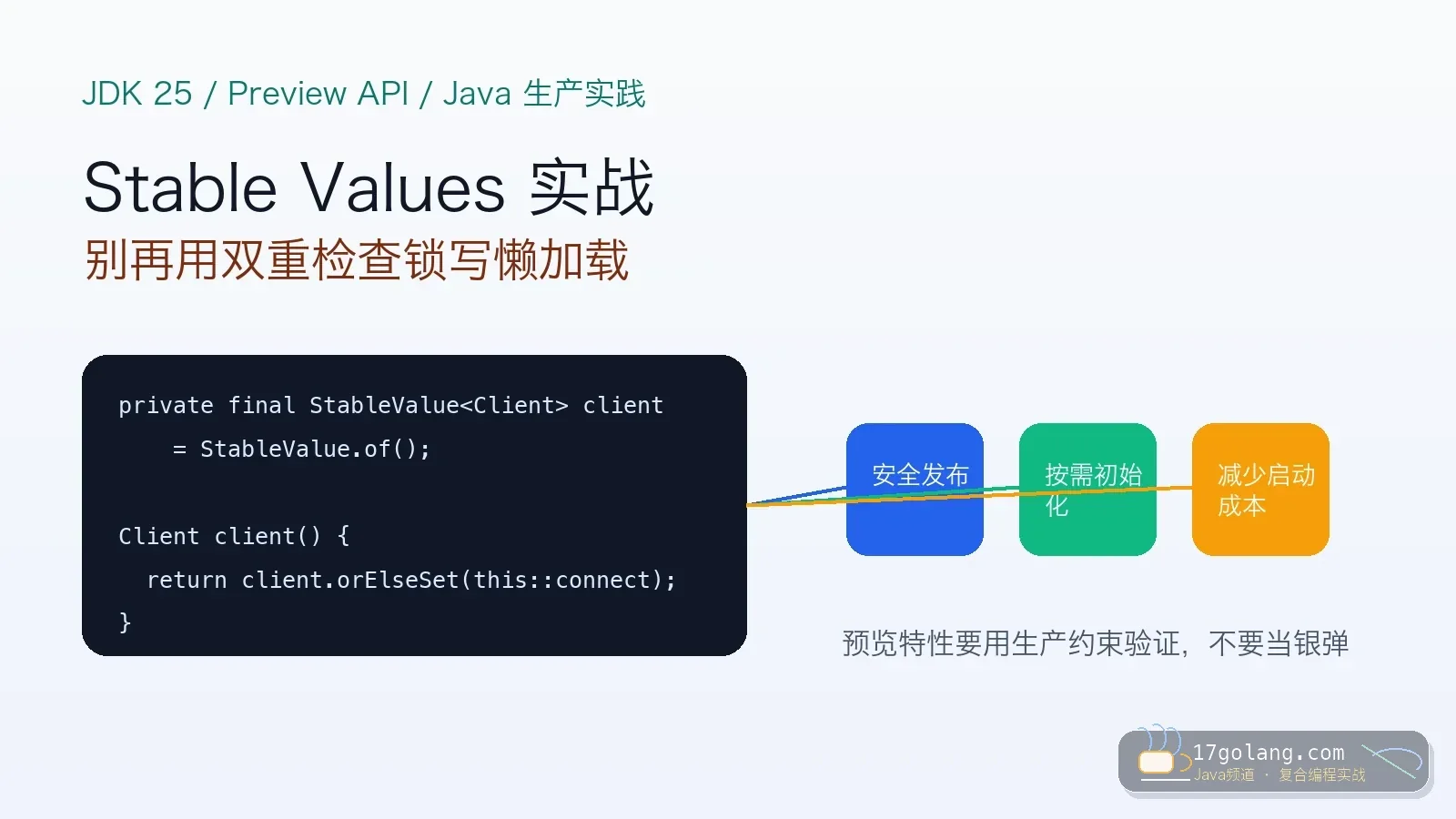
- 文章 · java教程 | 6天前 | 并发编程 · 生产实践 · Java教程 · JDK25 · 虚拟线程 · 虚拟线程 Java 25 JEP 505 Structured Concurrency StructuredTaskScope
- Java 25 Structured Concurrency 实战:别让 CompletableFuture 把超时拖散
- 443浏览 收藏
-
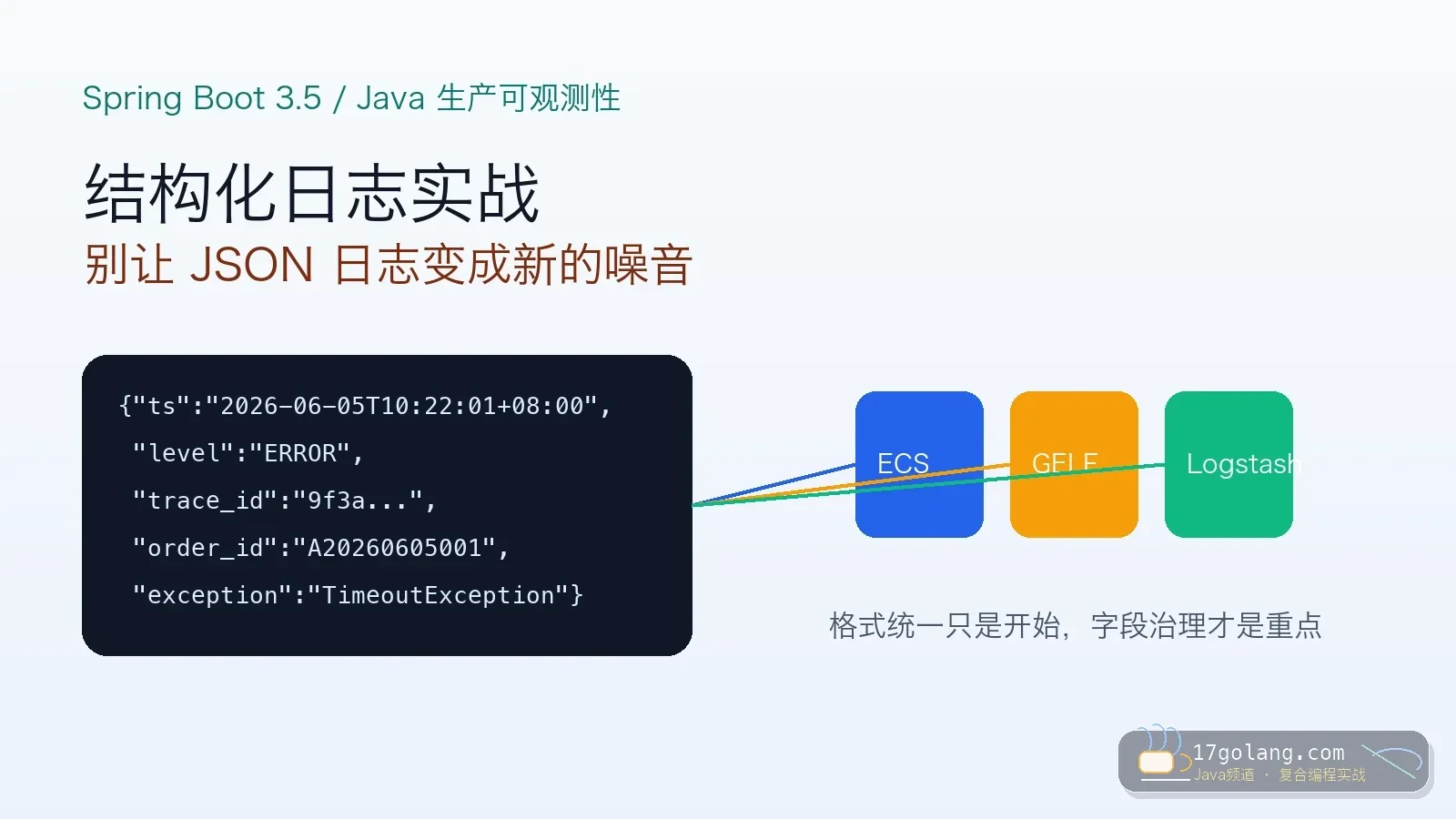
- 文章 · java教程 | 1星期前 | 日志 · Spring Boot · 生产实践 · 可观测性 · Java教程 · java 可观测性 MDC 结构化日志 Spring Boot 3.5
- Spring Boot 3.5 结构化日志实战:别让 JSON 日志变成新的噪音
- 332浏览 收藏
-
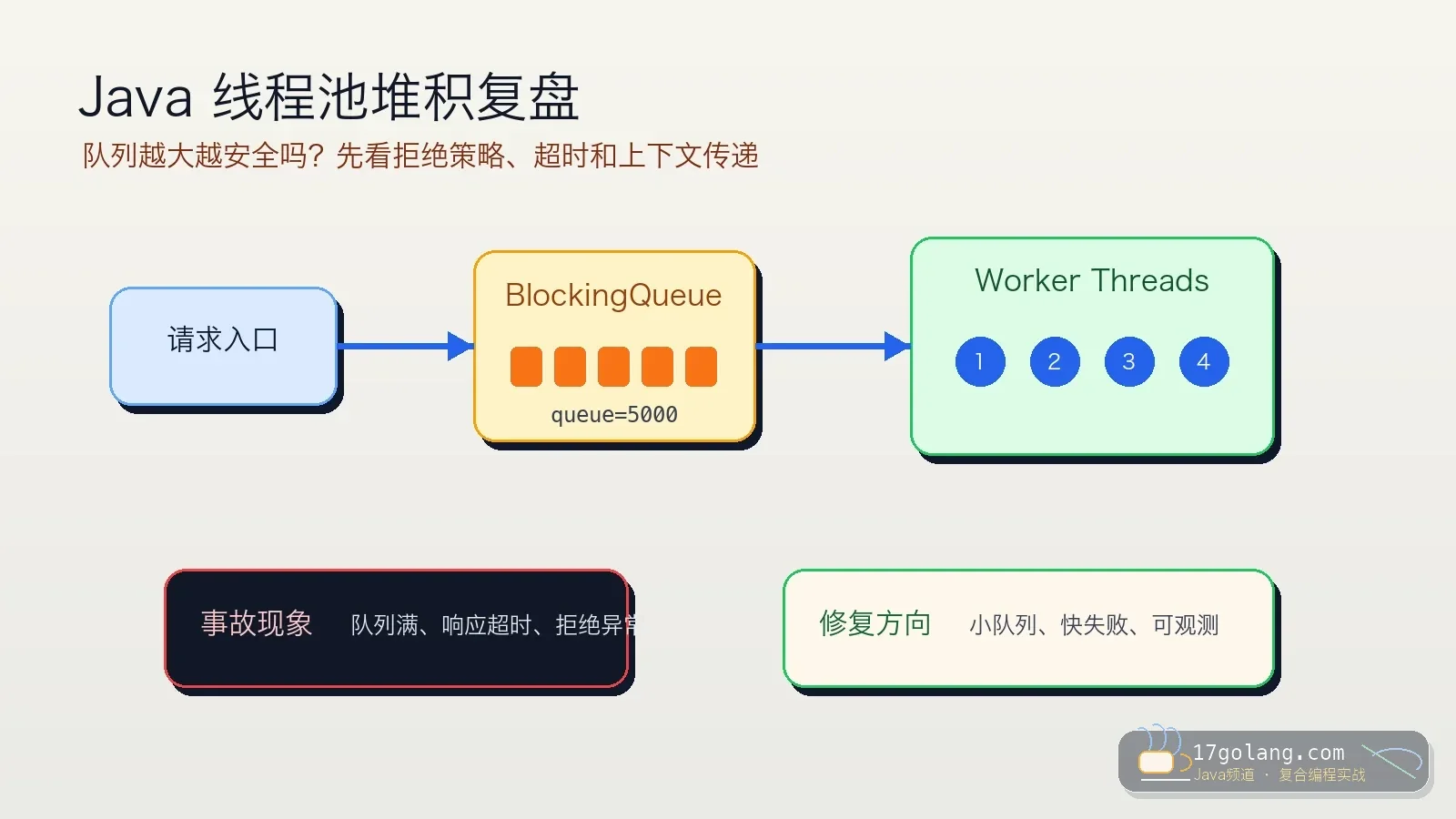
- 文章 · java教程 | 1星期前 | 线程池 · Spring Boot · 生产实践 · Java教程 · ThreadPoolExecutor · java 性能优化 线程池 spring boot threadpoolexecutor
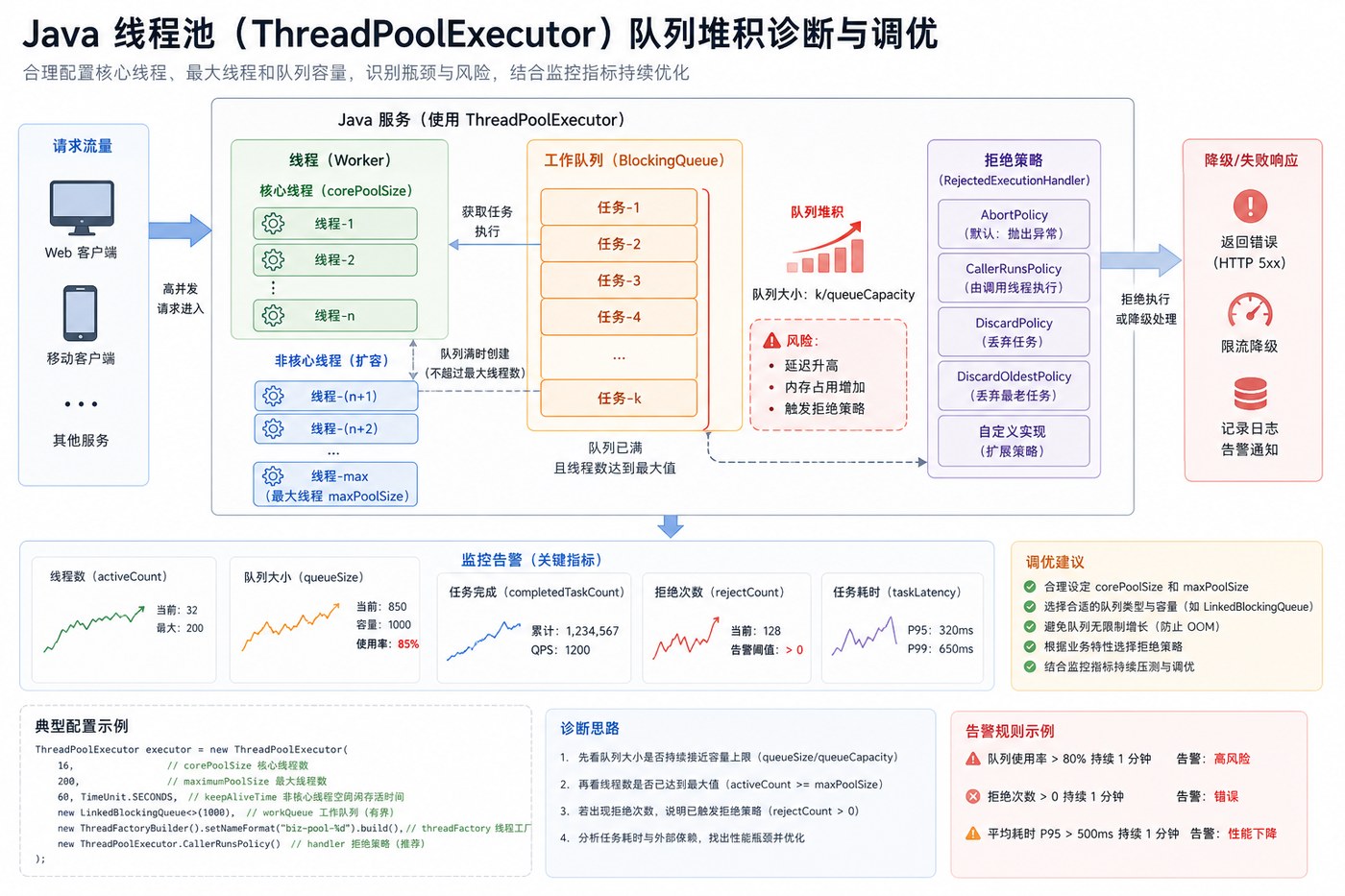
- Java 线程池队列堆积复盘:别让无界队列把慢故障藏起来
- 326浏览 收藏

-

- 前端进阶之JavaScript设计模式
- 设计模式是开发人员在软件开发过程中面临一般问题时的解决方案,代表了最佳的实践。本课程的主打内容包括JS常见设计模式以及具体应用场景,打造一站式知识长龙服务,适合有JS基础的同学学习。
- 543次学习
-

- GO语言核心编程课程
- 本课程采用真实案例,全面具体可落地,从理论到实践,一步一步将GO核心编程技术、编程思想、底层实现融会贯通,使学习者贴近时代脉搏,做IT互联网时代的弄潮儿。
- 516次学习
-

- 简单聊聊mysql8与网络通信
- 如有问题加微信:Le-studyg;在课程中,我们将首先介绍MySQL8的新特性,包括性能优化、安全增强、新数据类型等,帮助学生快速熟悉MySQL8的最新功能。接着,我们将深入解析MySQL的网络通信机制,包括协议、连接管理、数据传输等,让
- 500次学习
-

- JavaScript正则表达式基础与实战
- 在任何一门编程语言中,正则表达式,都是一项重要的知识,它提供了高效的字符串匹配与捕获机制,可以极大的简化程序设计。
- 487次学习
-

- 从零制作响应式网站—Grid布局
- 本系列教程将展示从零制作一个假想的网络科技公司官网,分为导航,轮播,关于我们,成功案例,服务流程,团队介绍,数据部分,公司动态,底部信息等内容区块。网站整体采用CSSGrid布局,支持响应式,有流畅过渡和展现动画。
- 485次学习
-
- ChatExcel酷表
- ChatExcel酷表是由北京大学团队打造的Excel聊天机器人,用自然语言操控表格,简化数据处理,告别繁琐操作,提升工作效率!适用于学生、上班族及政府人员。
- 8609次使用
-
- Any绘本
- 探索Any绘本(anypicturebook.com/zh),一款开源免费的AI绘本创作工具,基于Google Gemini与Flux AI模型,让您轻松创作个性化绘本。适用于家庭、教育、创作等多种场景,零门槛,高自由度,技术透明,本地可控。
- 9029次使用
-
- 可赞AI
- 可赞AI,AI驱动的办公可视化智能工具,助您轻松实现文本与可视化元素高效转化。无论是智能文档生成、多格式文本解析,还是一键生成专业图表、脑图、知识卡片,可赞AI都能让信息处理更清晰高效。覆盖数据汇报、会议纪要、内容营销等全场景,大幅提升办公效率,降低专业门槛,是您提升工作效率的得力助手。
- 8853次使用
-

- 星月写作
- 星月写作是国内首款聚焦中文网络小说创作的AI辅助工具,解决网文作者从构思到变现的全流程痛点。AI扫榜、专属模板、全链路适配,助力新人快速上手,资深作者效率倍增。
- 10755次使用
-
- MagicLight
- MagicLight.ai是全球首款叙事驱动型AI动画视频创作平台,专注于解决从故事想法到完整动画的全流程痛点。它通过自研AI模型,保障角色、风格、场景高度一致性,让零动画经验者也能高效产出专业级叙事内容。广泛适用于独立创作者、动画工作室、教育机构及企业营销,助您轻松实现创意落地与商业化。
- 9693次使用
-
- 提升Java功能开发效率的有力工具:微服务架构
- 2023-10-06 501浏览
-
- 掌握Java海康SDK二次开发的必备技巧
- 2023-10-01 501浏览
-
- 如何使用java实现桶排序算法
- 2023-10-03 501浏览
-
- Java开发实战经验:如何优化开发逻辑
- 2023-10-31 501浏览
-
- 如何使用Java中的Math.max()方法比较两个数的大小?
- 2023-11-18 501浏览


